![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
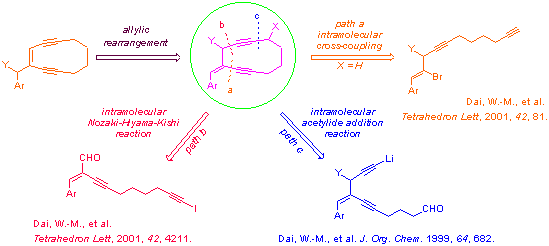
Enediynes are a novel class of antitumor antibiotics possessing complex molecular structures and intriguing biological reaction mechanisms (Nicolaou, K. C.; Dai, W.-M. Angew. Chem., Int. Ed. Engl. 1991, 30, 1387). Over the past years, we have developed an allylic rearrangement approach to the formation of conjugated (Z)-hexa-1,5-diyn-3-ene structures with high regio- and stereoselectivity (Dai, W.-M., et al. Angew. Chem., Int. Ed. Engl. 1996, 35, 779; Tetrahedron Lett. 1996, 37, 8413; Tetrahedron Lett. 1998, 39, 8149; J. Org. Chem. 1999, 64, 5062). This methodology provides us a very useful tool for designing and synthesis of cyclic enediyne prodrugs with improved chemical stability before activation of the enediyne reaction cascade. We have now developed three ring closure reactions for the synthesis of a small library of (E)-4-(arylmethylidene)cyclodeca-1,5-diyne-3-ols and their ester derivatives. These include: an intramolecular Sonogashira cross-coupling reaction (path a), an intramolecular Nozaki-Hiyama-Kishi reaction (path b), and an intramolecular acetylide addition reaction (path c). We have engineered different aromatic rings (Ar) for facilitating the allylic rearrangement. Preliminary biological screening on the DNA cleavage potency and in vitro anticancer activity has revealed that this class of synthetic enediyne prodrugs is the promising lead structure for further development.
![]()
Atropisomerism is a
phenomenon associated
with the restricted rotation around a single bond. The binaphthyl derivatives
are the classical examples of atropisomers and they have been extensively used
as the chiral auxiliaries and ligands in asymmetric synthesis and catalysis.
Recently, non-biaryl atropisomers such as the axially chiral amides
have
received considerable attention on the applications as chiral reagents,
auxiliaries, and ligands. These amides cover the anilides, the N-arylimides,
the benzamides, and the 1-naphthamides. Moreover, other axially chiral
C1-substituted naphthalenes, for example, the C1-vinyl naphthalenes (used in
enantioselective alkylation), the C1-arylsulfinyl naphthalenes, and the
2-methyl-1-naphthylphosphine oxide were also explored.
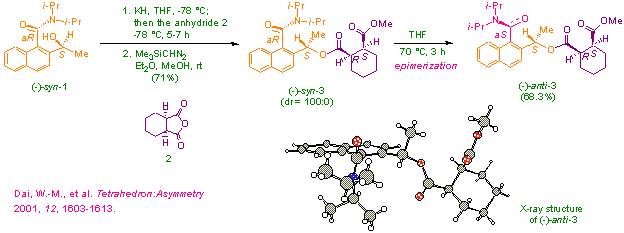
We
have prepared enantiomerically pure 2-substituted 1-naphthamides such as (–)-syn-1
via resolution
over a chiral stationary phase and determined the absolute stereochemistry.
Synthesis of these axially chiral 2-substituted 1-naphthamides through
asymmetric reactions is under active investigation. We have used (–)-syn-1
in enantioselective
desymmetrization of cyclic meso anhydrides such as 2 to provide
the product (–)-syn-3
with 100% diastereoselectivity.
![]()
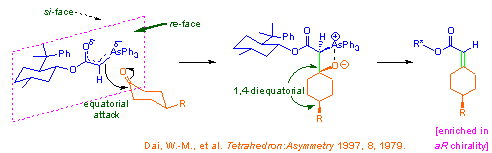
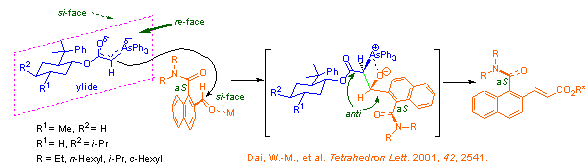
Arsonium ylides are known to be more reactive than the corresponding phosphonium analogs. For example, the arsonium ylides stabilized by an electron-withdrawing group react with aldehydes and ketones below room temperature. We took this reactivity advantage and used the menthol-derived arsonium ylides in asymmetric desymmetrization of 4-substituted cyclohexanones and in a kinetic resolution of axially chiral N,N-dialkyl-2-formyl-1-naphthamides. Synthesis of chiral arsine compounds and their asymmetric reactions are currently under active investigation.
Asymmetric desymmetrization of 4-substituted cyclohexanones with up to 91:9 diastereomeric ratio:
Kinetic resolution of axially chiral N,N-dialkyl-2-formyl-1-naphthamides with up to 88:12 diastereomeric ratio:
![]()
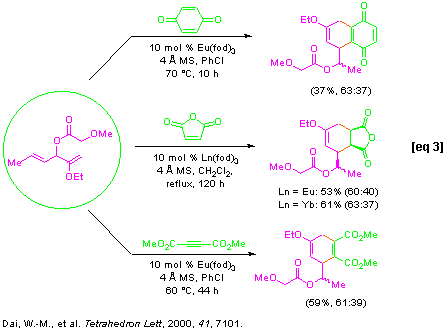
Eu(fod)3 was demonstrated to catalyze rearrangement of allylic methoxyacetates under mild reaction conditions (Shull, B. K.; Sakai, T.; Koreeda, M. J. Am. Chem. Soc. 1996, 118, 11690). We found that the lanthanide (III) complexes, Ln(fod)3 and Ln(hfc)3 with Ln = Er, Eu, Pr and Yb selectively catalyzed rearrangements of allylic alkoxyacetates at room temperature. We have successfully applied this allylic rearrangement to the synthesis of 10-membered ring enediynes [eqs 1-2] and have established a tandem regiospecific rearrangement of divinyl alkoxyacetates and Diels-Alder reaction sequence for a ready access to the highly functionalized bicyclic molecules [eq 3].
Synthesis of 10-membered ring enediynes under mild conditions:
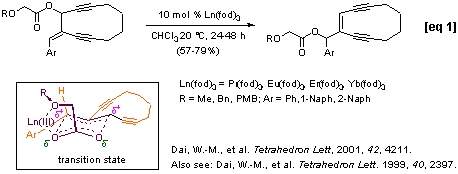
Intramolecular nucleophilic group assisted rearrangement of allylic esters:
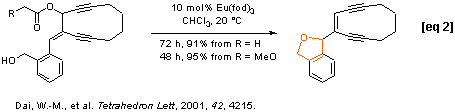
Tandem regiospecific rearrangement of divinyl alkoxyacetates and Diels-Alder reaction:
![]()
Organic synthesis on solid supports is a rapidly growing research area and serves as one of the enabling technologies for combinatorial library synthesis. Our interest in this area focuses on the solid phase metal-catalyzed cross-coupling reactions and application to the construction of bioactive heterocyclic compound libraries. We have established a novel synthesis of indoles from 2-aminophenols using the modified Sonogashira cross-coupling of 2-carboxamidoaryl triflates with 1-alkynes as the key step. Solid phase synthesis of indole compound libraries starting from 2-aminophenols based on our chemistry is currently under active investigation.
Novel synthesis of indoles from 2-aminophenols:
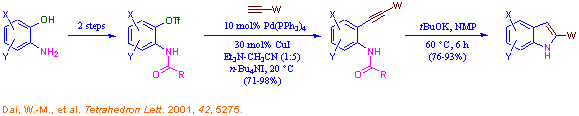
![]()
![]()
![]()
![]()
![]()